Viewing BAM alignment files | Private annotations

|
|
Table of Contents
: Data visualization
: Viewing BAM alignment files | Private annotations |
|
Alamut® Visual BAM viewer main features:
In this section:
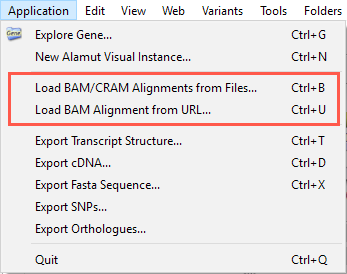
Two options are available to load alignment files:
Using CRAM files usually requires defining the location of reference sequences. See CRAM settings

 Sequencing targets
Sequencing targets Depth of coverage
Depth of coverage Reads
Reads Variants
VariantsWhen zooming at nucleotide level the genomic reference sequence is displayed above the targets sub-track:
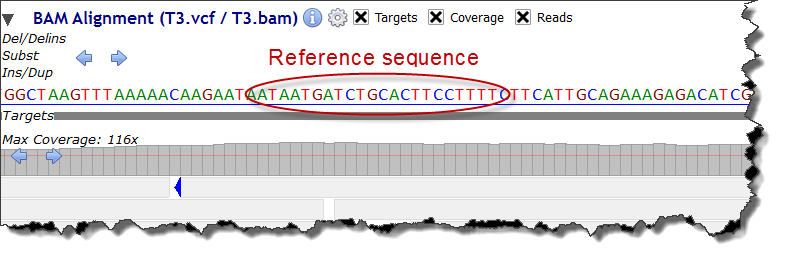
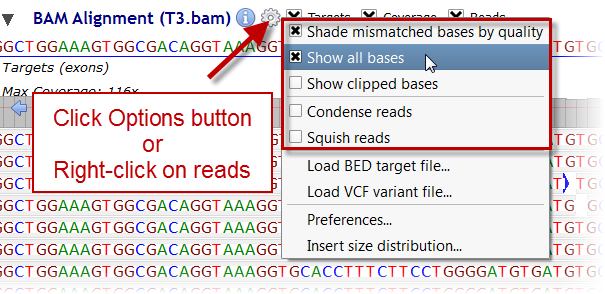
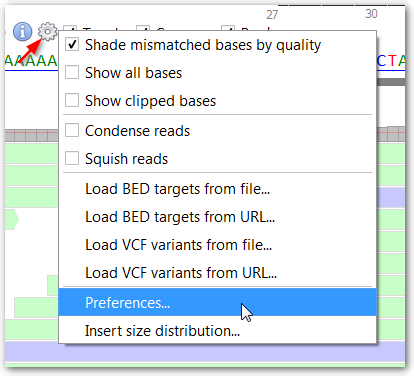
By default, sequencing targets are supposed to cover current gene's exons.
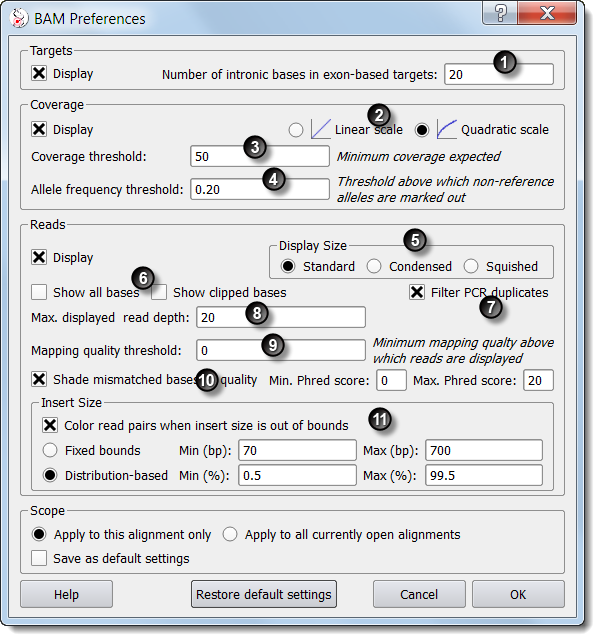
This setting specifies the number of exon-flanking intronic bases to add to exon-defined targets.
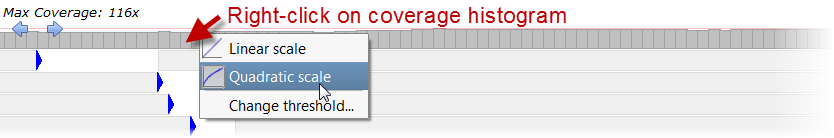
(Targets can otherwise be loaded from BED files using "Load BED target file..." in the BAM track Options menu  ). The coverage histogram can either be displayed using a linear
scale where the height of each bar is directly proportionate to the depth value, or using a quadratic scale where low depth
values are increased and high depth values are decreased.
Targets are highlighted with red color where coverage depth is below this threshold. Beside, detected SNVs are only reported at positions where coverage is above this threshold. Single nucleotide variants (SNVs) are detected where non-reference bases are called at a frequency above this threshold.
). The coverage histogram can either be displayed using a linear
scale where the height of each bar is directly proportionate to the depth value, or using a quadratic scale where low depth
values are increased and high depth values are decreased.
Targets are highlighted with red color where coverage depth is below this threshold. Beside, detected SNVs are only reported at positions where coverage is above this threshold. Single nucleotide variants (SNVs) are detected where non-reference bases are called at a frequency above this threshold. These settings define the graphical height of reads.
These settings define the graphical height of reads. If 'Show all bases' is not checked then read bases are displayed
only if they differ from the reference sequence. Some NGS data processing tools "soft-clip" bases at either end of reads if appropriate. Checking the 'Show clipped bases' option reveals these bases.
This option should usually remain unchecked.
If 'Show all bases' is not checked then read bases are displayed
only if they differ from the reference sequence. Some NGS data processing tools "soft-clip" bases at either end of reads if appropriate. Checking the 'Show clipped bases' option reveals these bases.
This option should usually remain unchecked. Alamut® Visual does not itself detect PCR duplicates. Reads marked
as PCR duplicates in the BAM file are withdrawn if this option is checked.
Alamut® Visual does not itself detect PCR duplicates. Reads marked
as PCR duplicates in the BAM file are withdrawn if this option is checked.
 This setting only affects the graphical display of reads, not computations.
This setting only affects the graphical display of reads, not computations. Reads with a mapping quality under this threshold are withdrawn.
Reads with a mapping quality under this threshold are withdrawn. If this option is checked then called bases with a Phred score
under the specified minimum threshold are not displayed. Bases with a Phred score between the specified minimum and maximum thresholds are shaded.
Bases above the maximum threshold are displayed in full color.
If this option is checked then called bases with a Phred score
under the specified minimum threshold are not displayed. Bases with a Phred score between the specified minimum and maximum thresholds are shaded.
Bases above the maximum threshold are displayed in full color.
 Based on insert size values provided in the BAM file, paired reads too distant or too close from
each other are highlighted if this option is checked. Expected normal insert sizes can be expressed as fixed values or as a percentage over the distribution.
If the insert size is large, denoting a deletion, reads are colored red. If the insert size is small, denoting an insertion, reads are colored blue.
Based on insert size values provided in the BAM file, paired reads too distant or too close from
each other are highlighted if this option is checked. Expected normal insert sizes can be expressed as fixed values or as a percentage over the distribution.
If the insert size is large, denoting a deletion, reads are colored red. If the insert size is small, denoting an insertion, reads are colored blue.
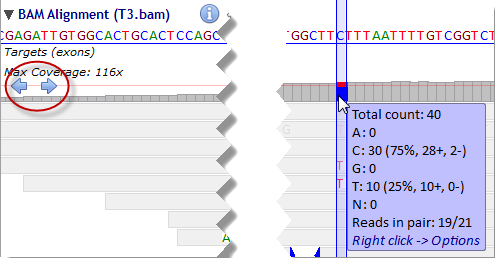
Single nucleotide variants (SNVs) are automatically detected where non-reference bases are called at a frequency above the 'Allele frequency threshold' defined in the Preferences panel. Jump from one SNV to the other by clicking the arrow buttons on the left of the coverage sub-track:
You can also load called variants from VCF files and, now, gVCF files.
You have two possibilities to load VCF files:
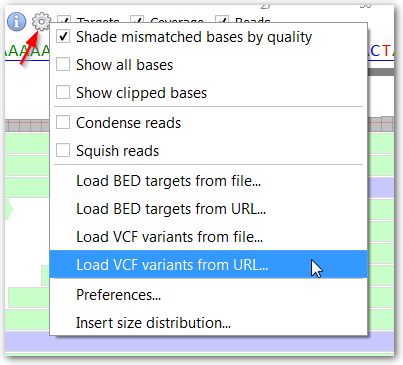
This creates a sub-track above the reference sequence. Jump from one variant to the other by clicking the arrow buttons on the left.

In Alamut® Visual, sequencing targets are supposed to cover current gene's exons with some exon-flanking intronic bases (20bp, by default).
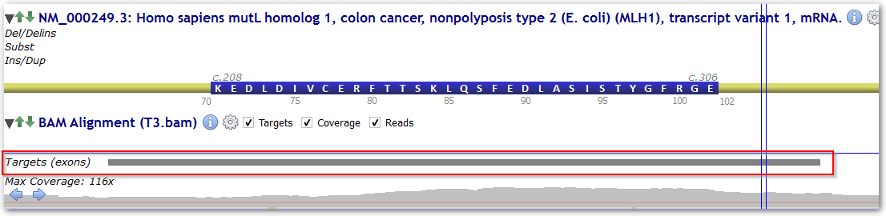
You can also load targeted regions from BED files in the BAM track. You have two possibilities to load BED files:
> 'Options':

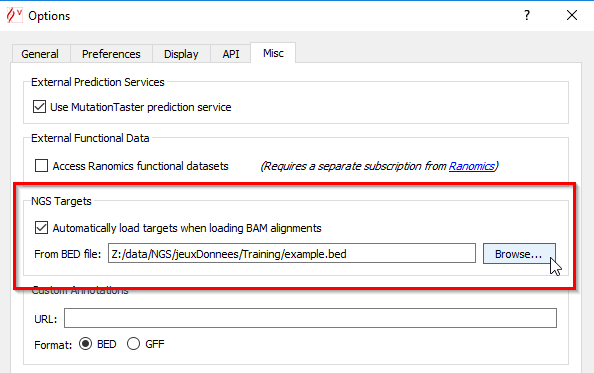
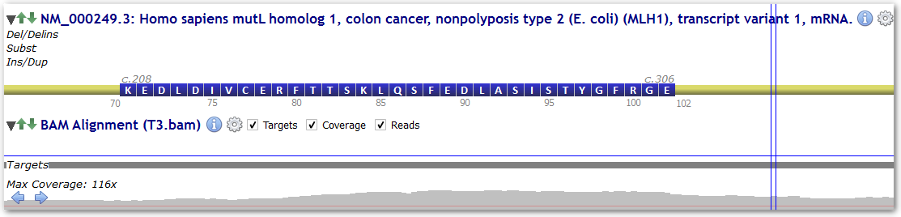
The sub-track named "Targets" is updated with your BED defined targets.
Alamut® Visual computes descriptive statistics from BAM file (depth of coverage and insert size). These statistics are computed for the current displayed gene locus.
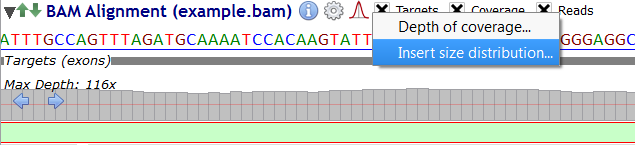
Click on the picture and, then select either "Depth of Coverage" or "Insert Size Distribution" from the menu.
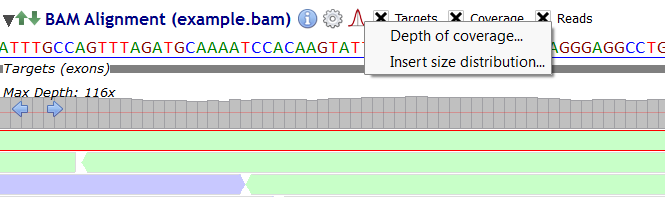
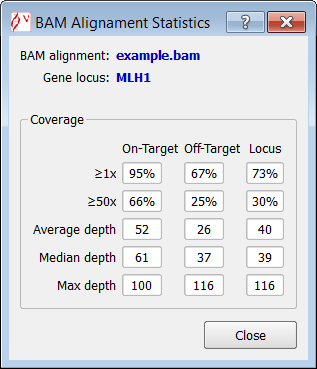
To display the depth of coverage statistics, select "Depth of Coverage" from the menu:

A table summarizes for the current gene locus, the following statistics:
To display the insert size distribution, select "Insert Size Distribution" from the menu:
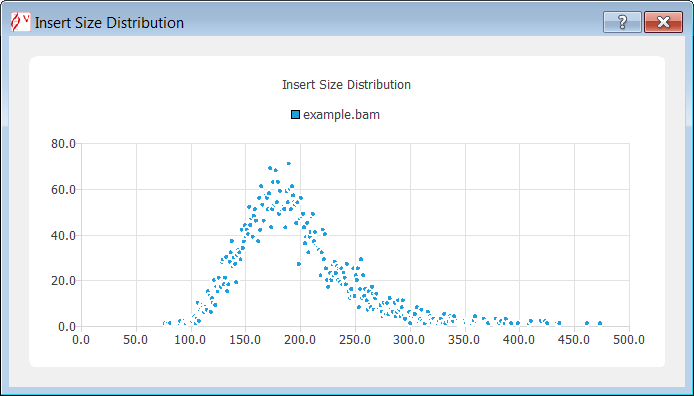
The insert sizes are computed from the reads that are in the current gene locus (displayed in Alamut® Visual) and, then they are plotted as follows:
This section is rather technical. Help from bioinformaticians could be beneficial to define appropriate CRAM settings.
CRAM handling in Alamut is based on Samtools.
As explained here Samtools needs the reference genome sequence in order to decode a CRAM file. It uses the MD5 sum of each reference sequence as the key to link a CRAM file to the reference genome used to generate it (see also the Samtools man page).
Samtools can use either the REF_PATH or REF_CACHE environment variables to find reference sequences. Input fields for these variables are available in menu Tools > Options > Misc tab: CRAM Settings, as shown below.
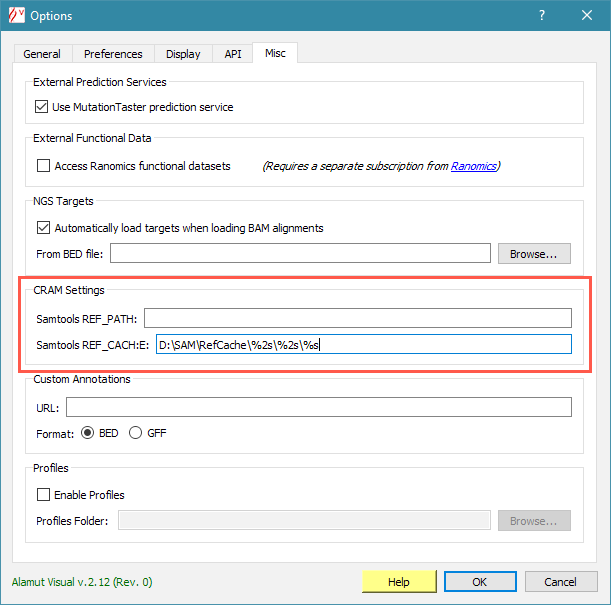
You will need to provide the path to MD5 reference sequences in one of these fields (unless you use CRAM files with embedded reference sequences).
For your convenience we have prepared a package of reference MD5 files for GRCh37 and GRCh38 primary sequences. It is available here:
http://downloads.interactive-biosoftware.com/CRAM/RefCache.tgz (uncompress and untar this file)
If you use this MD5 reference directory and its path in your filesystem is /somepath/RefCache then in the 'Samtools REF_CACHE' field of the Alamut Options box enter: /somepath/RefCache/%2s/%2s/%s
© 2020 Interactive Biosoftware - Last modified: 30 May 2019